
Sentieon | Application Tutorial: Germline Variant Detection Analysis of HiFi Long-Read Data Using DNAscope

Introduction
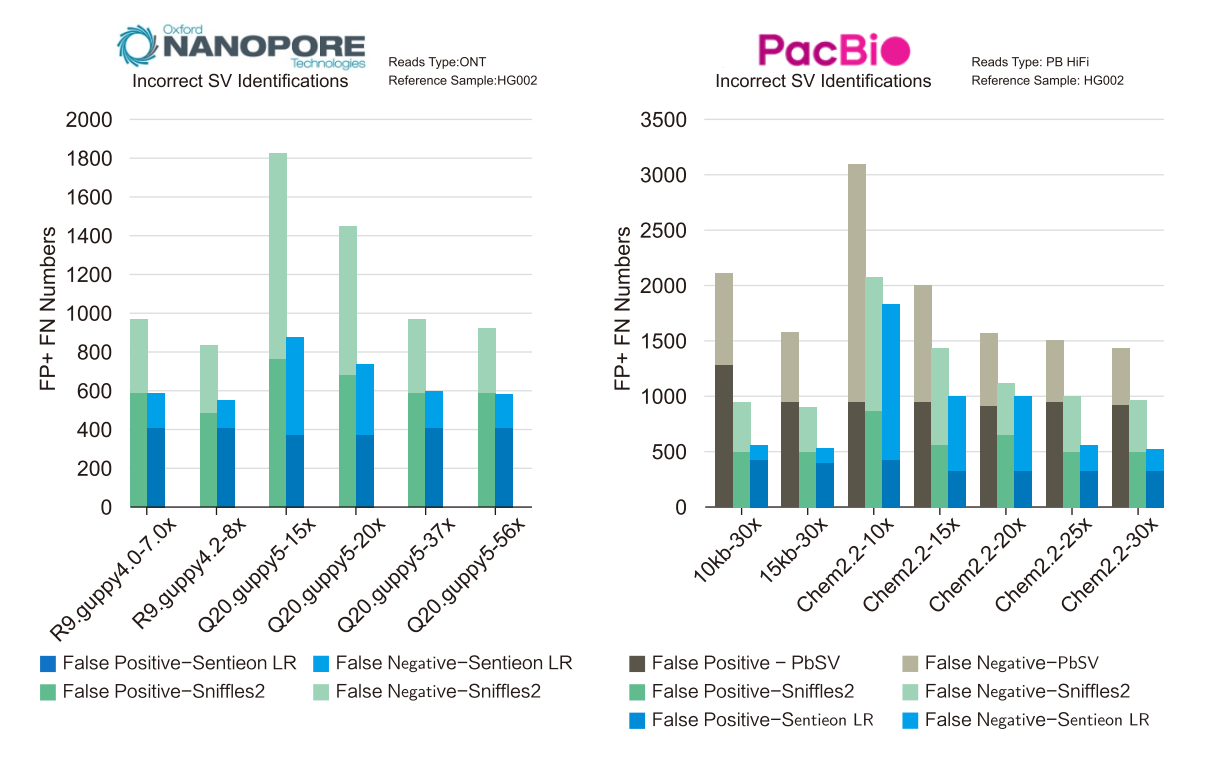
This document describes germline variant calling for PacBio® HiFi data using Sentieon® DNAscope. PacBio® HiFi technology produces high-quality long reads with quality scores above Q20 and average lengths between 10-25kb. These accurate long reads enable precise variant detection in genomic repeat regions that are challenging for short-read and noisy long-read methods.
Sentieon® DNAscope leverages the high quality and long read length advantages of PacBio® HiFi data, using a calibrated machine learning model for fast and accurate variant calling. The DNAscope pipeline for HiFi data takes aligned HiFi data as input and outputs variant calls in VCF format.
This pipeline requires Sentieon software version 202010.03 or newer and related scripts available from Sentieon®. Python version >2.7 or >3.3 and bcftools version 1.10 or higher are required. Python, bcftools, and bedtools must be in the user's environment variable.
Input Data Requirements
Aligned Data
The pipeline input uses PacBio® HiFi data aligned with pbmm2 or minimap2. For pbmm2 alignment, the recommended parameters are -c 0 -y 70 --preset HIFI. These settings disable pbmm2's traditional alignment consistency filtering, instead using gapped compressed sequence filtering and PacBio®'s recommended HiFi data alignment settings. For minimap2 alignment, the recommended parameter is -x map-hifi, which is minimap2's recommended setting for HiFi data.
Reference Genome
DNAscope will perform variant calling on the sample based on a high-quality reference genome. In addition to the reference genome, a genome index file (.fai) generated by samtools is required. We recommend using a reference genome without patch sequences.
Sentieon® DNAscope Pipeline for PacBio® HiFi Data
Pipeline Overview
This pipeline performs two rounds of variant calling, then merges the results to generate the final output. The specific steps are as follows:
-
The first round of calling detects variant sites in the sample;
-
Phasing is performed using the SNVs detected in the first round and long read information;
-
Second round of calling: In phased regions, variant calling is performed separately for each haplotype; In unphased regions, a more accurate diploid model is used for variant calling;
-
Variant sites from the first and second rounds are merged to generate the final result;
-
Special processing is applied to the MHC region using the provided MHC bed file to further improve variant detection accuracy;
The DNAscope machine learning model required for this pipeline can be obtained from https://github.com/Sentieon/sentieon-models.
Running the Pipeline
The HiFi data DNAscope pipeline can be run through a script containing multiple individual Sentieon commands. Variant detection and application of the machine learning model can be completed with a single command line. The HiFi data alignment file can be a bam or cram file aligned and indexed using pbmm2.
dnascope_HiFi.sh [-h] -r REFERENCE -i HIFI_BAM -m MODEL [-d dbSNP] [-B MHC_INTERVAL] [-b
INTERVAL] [-t NUMBER_THREADS] [-h] [--] VARIANT_VCFRequired parameters for the HiFi data Sentieon® DNAscope pipeline are:
-r REFERENCE: Path to the reference genome fasta file. Ensure that the reference genome file used is consistent with the one used in the alignment stage.
-i HIFI_BAM: Path to the aligned BAM file.
-m MODEL: DNAscope HiFi model file.
Optional parameters for the HiFi data Sentieon® DNAscope pipeline are:
-d dbSNP: Path to the dbSNP database VCF file. Only one file is needed. This file will be used for refSNP ID annotation in the variant detection results.
-B MHC_INTERVAL: MHC interval file in BED format. This file will be used for special processing of variant detection in the MHC region.
-b INTERVAL: Interval file in BED format. This file will restrict variant detection to the specified interval.
-t NUMBER_THREADS: Number of parallel threads. This parameter is optional; by default, all threads of the computer are used.
-h: Print help information.
Positional parameter for the HiFi data Sentieon® DNAscope pipeline:
VARIANT_VCF: Output filename for variant detection. This pipeline will output a bgzip compressed VCF file and its index file.
Pipeline Output Files
This pipeline will output a bgzip compressed file in VCF4.2 standard format (.vcf.gz) and its index file (.vcf.gz.tbi).
Other Considerations
Currently, this pipeline is only recommended for diploid samples. For samples containing both diploid and haploid regions, the -b INTERVAL parameter should be used to restrict variant calling to diploid chromosomes.